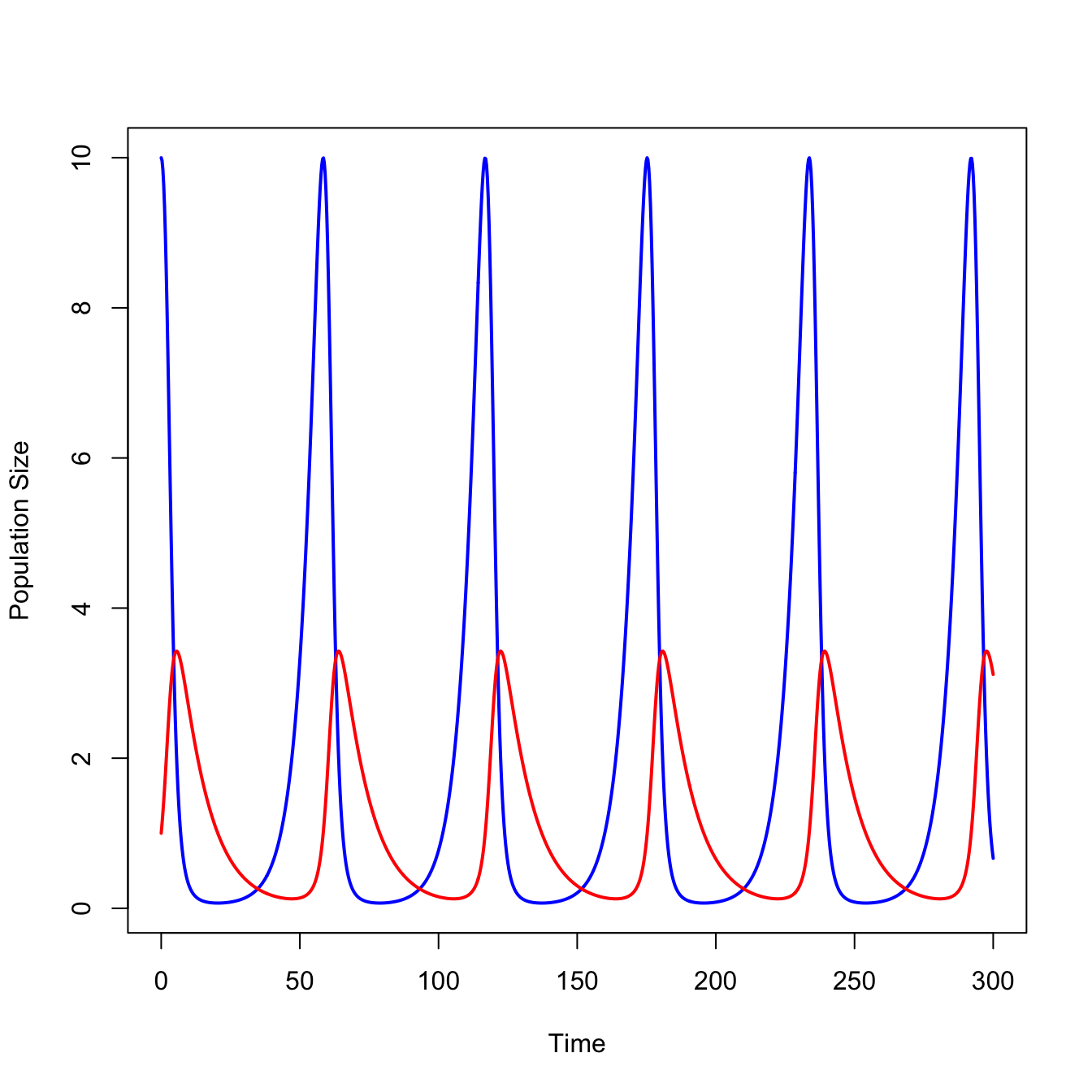
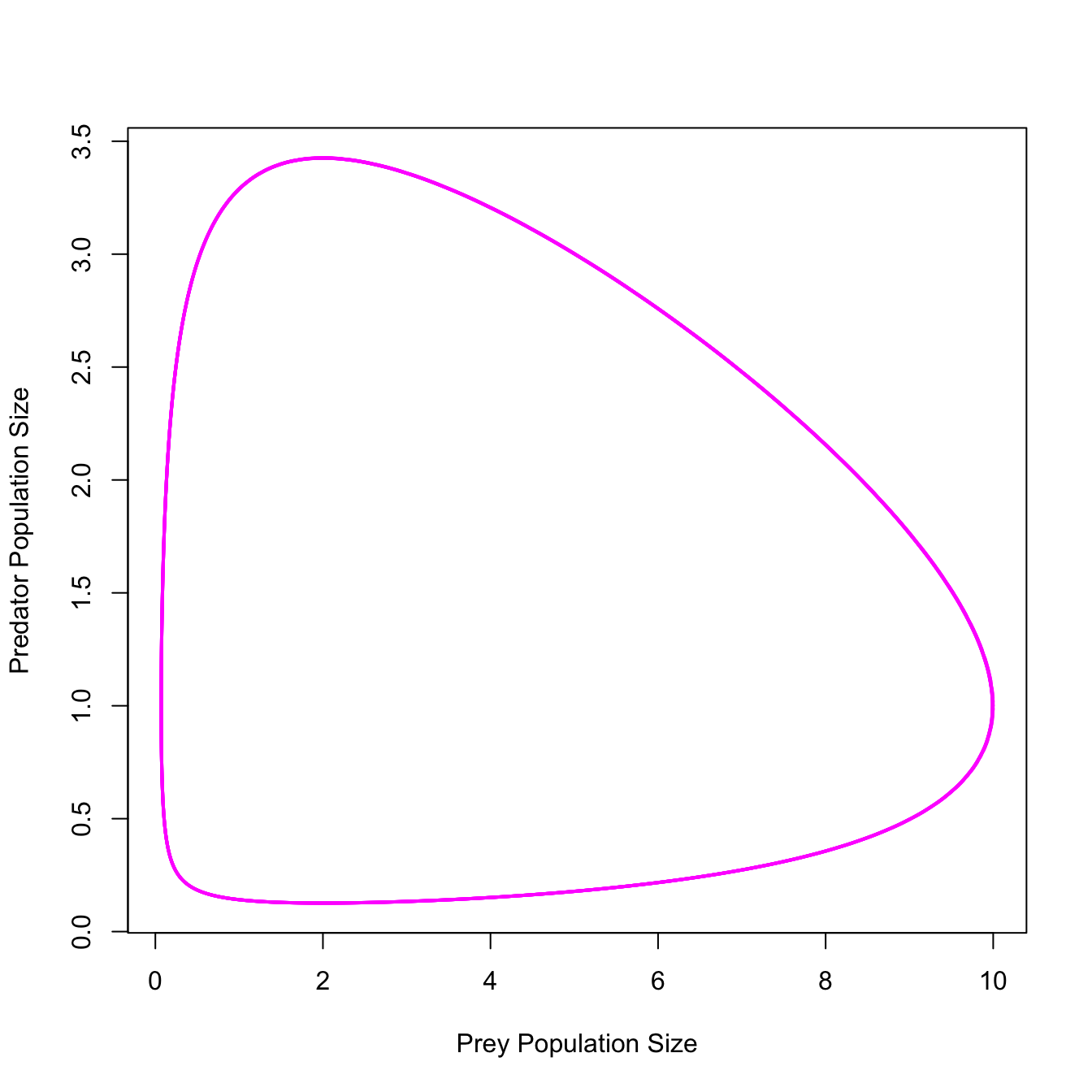
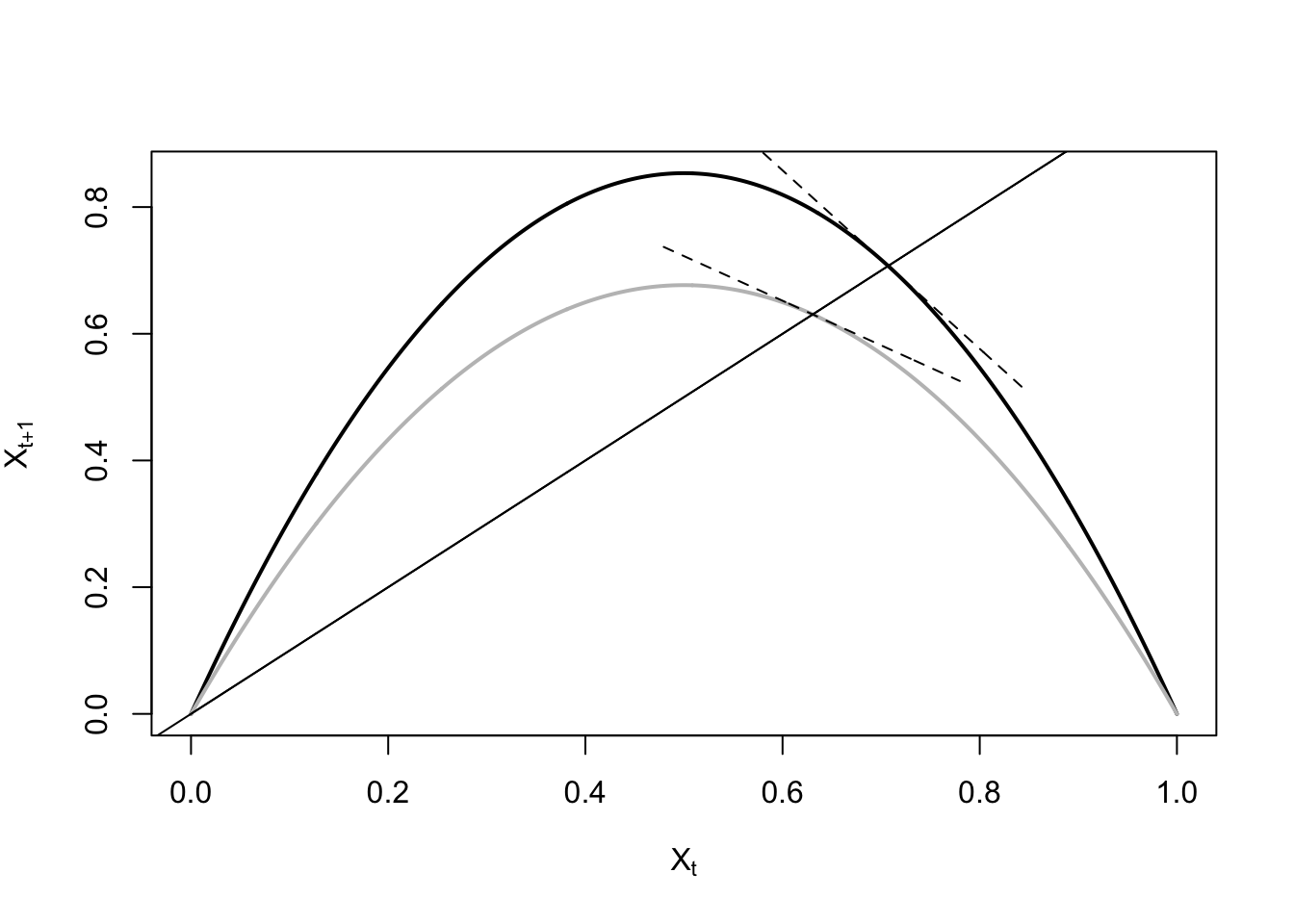
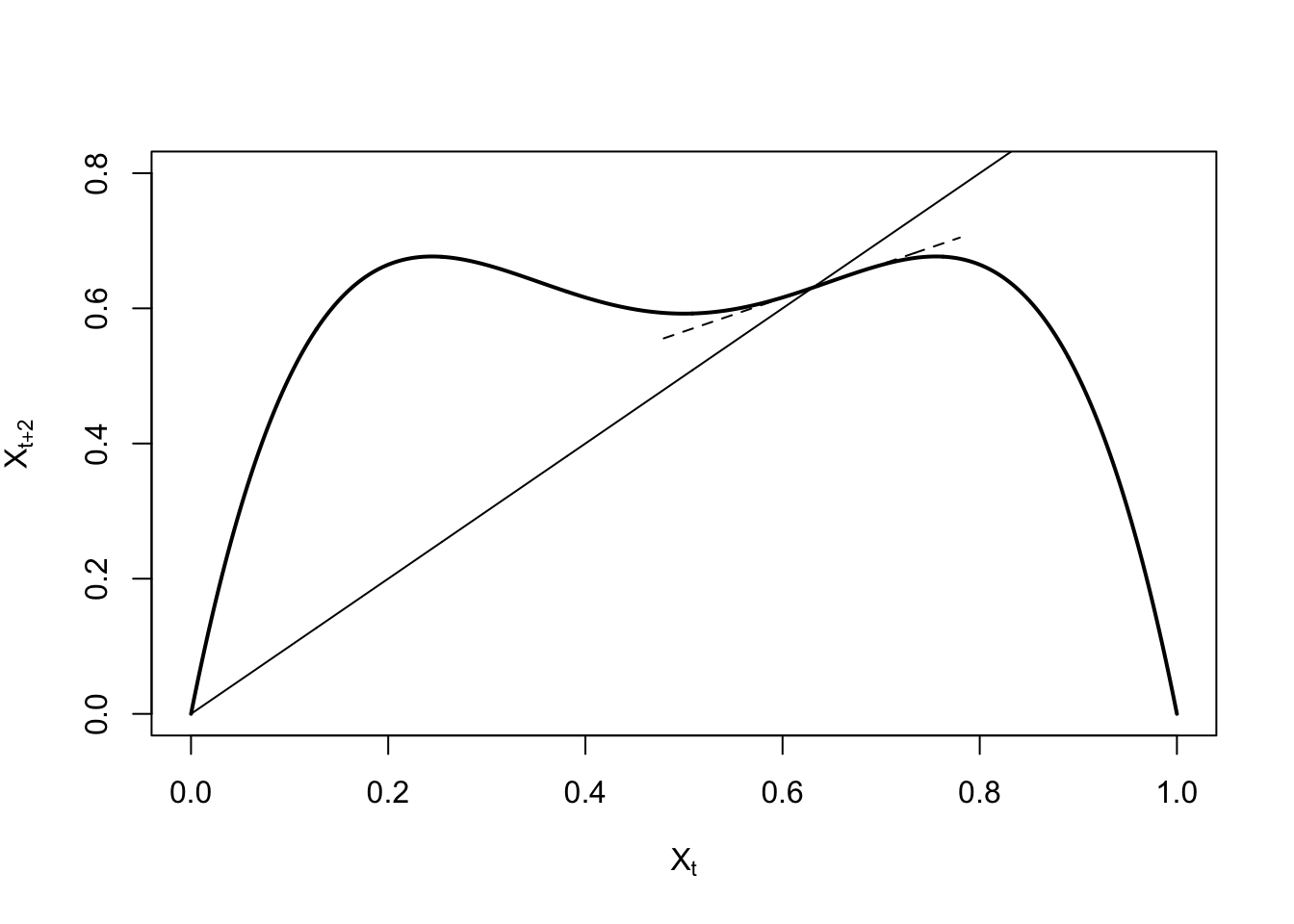
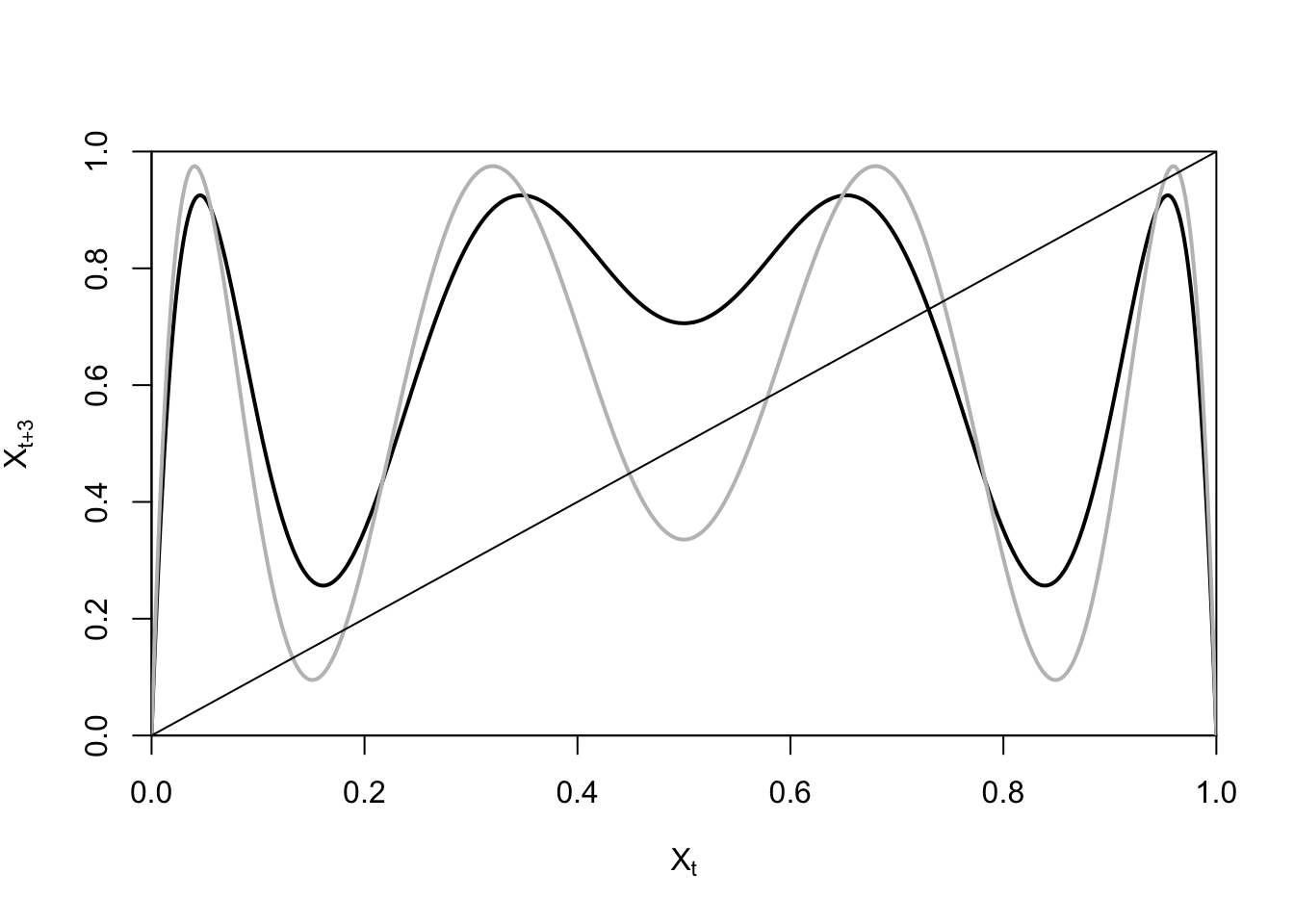
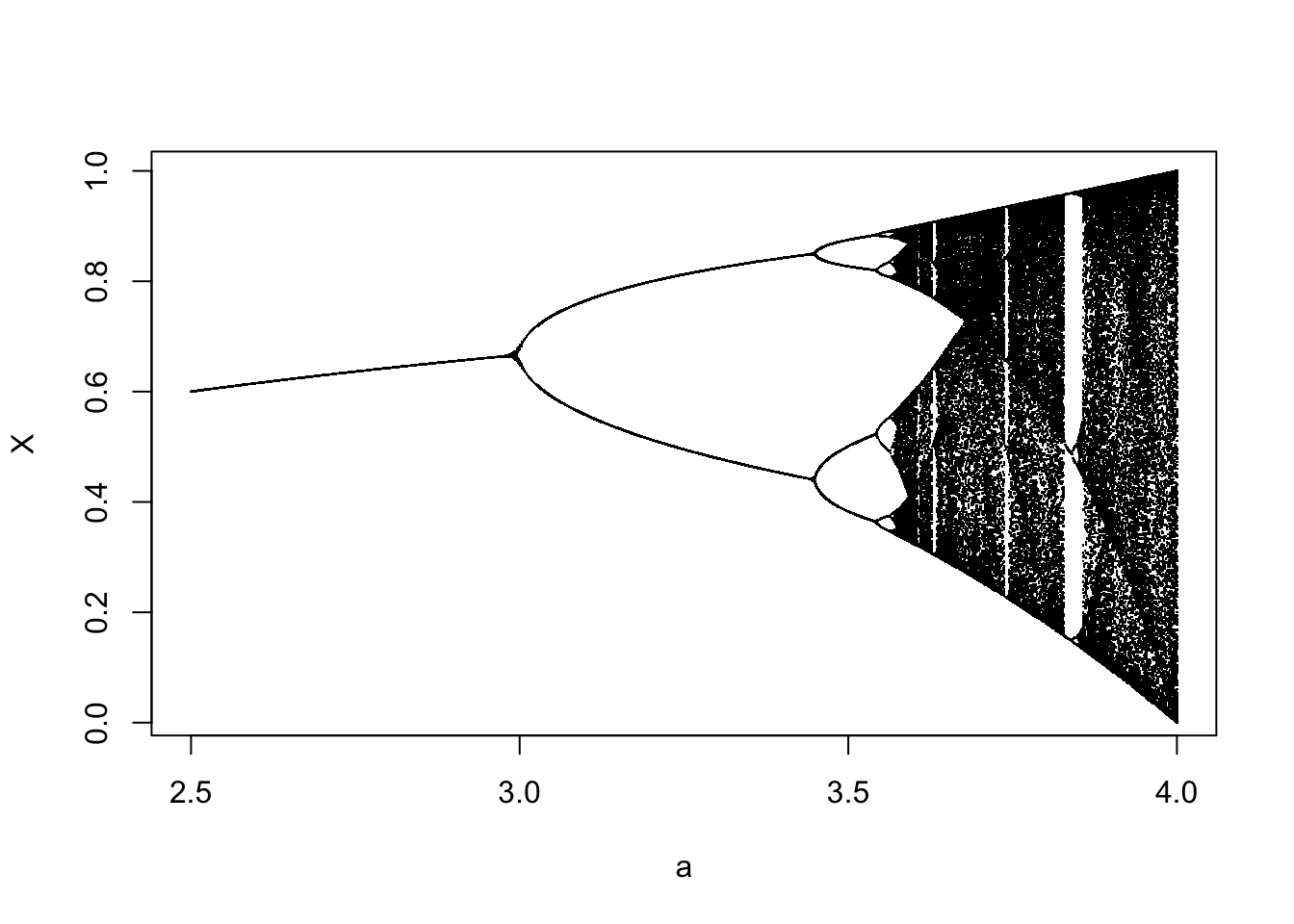
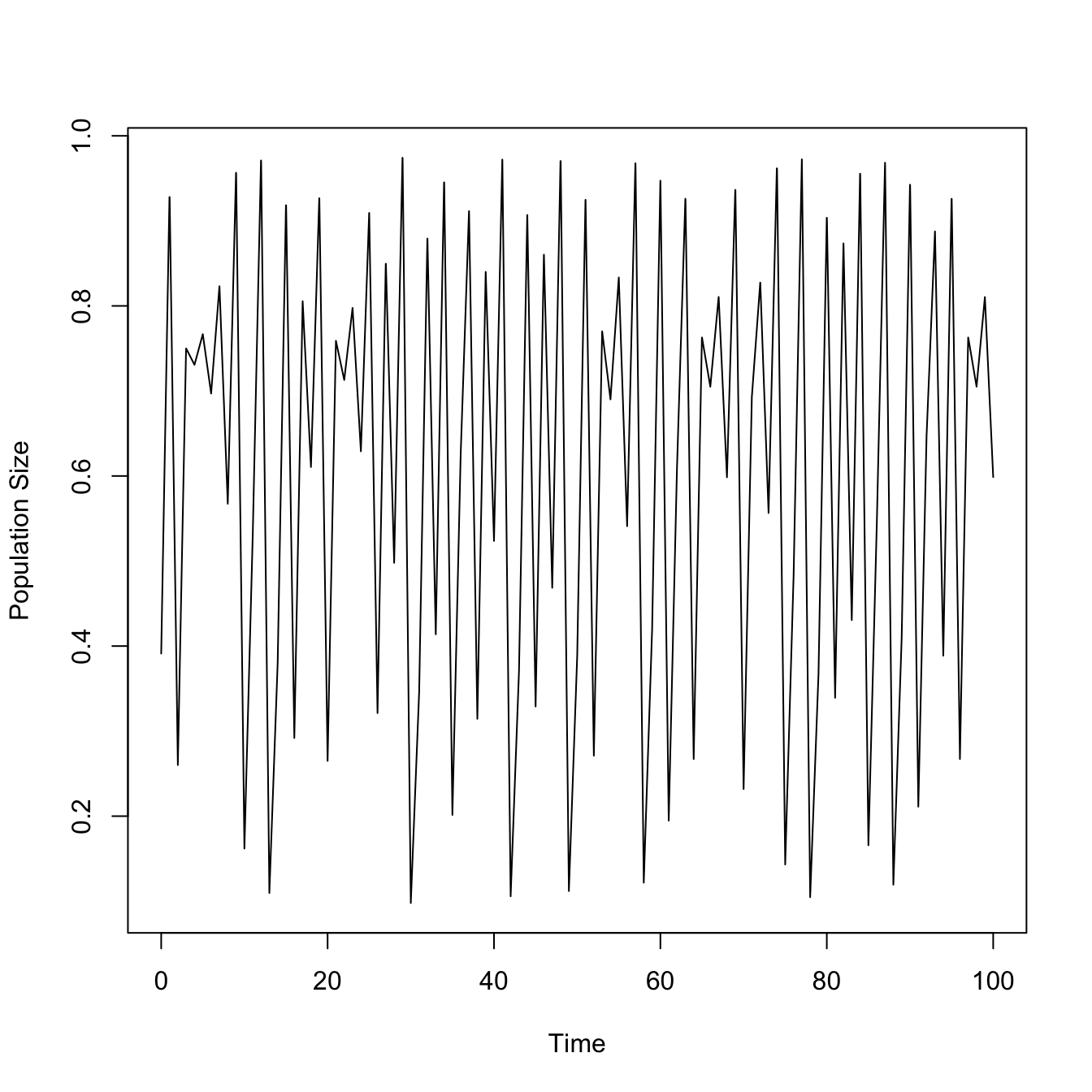
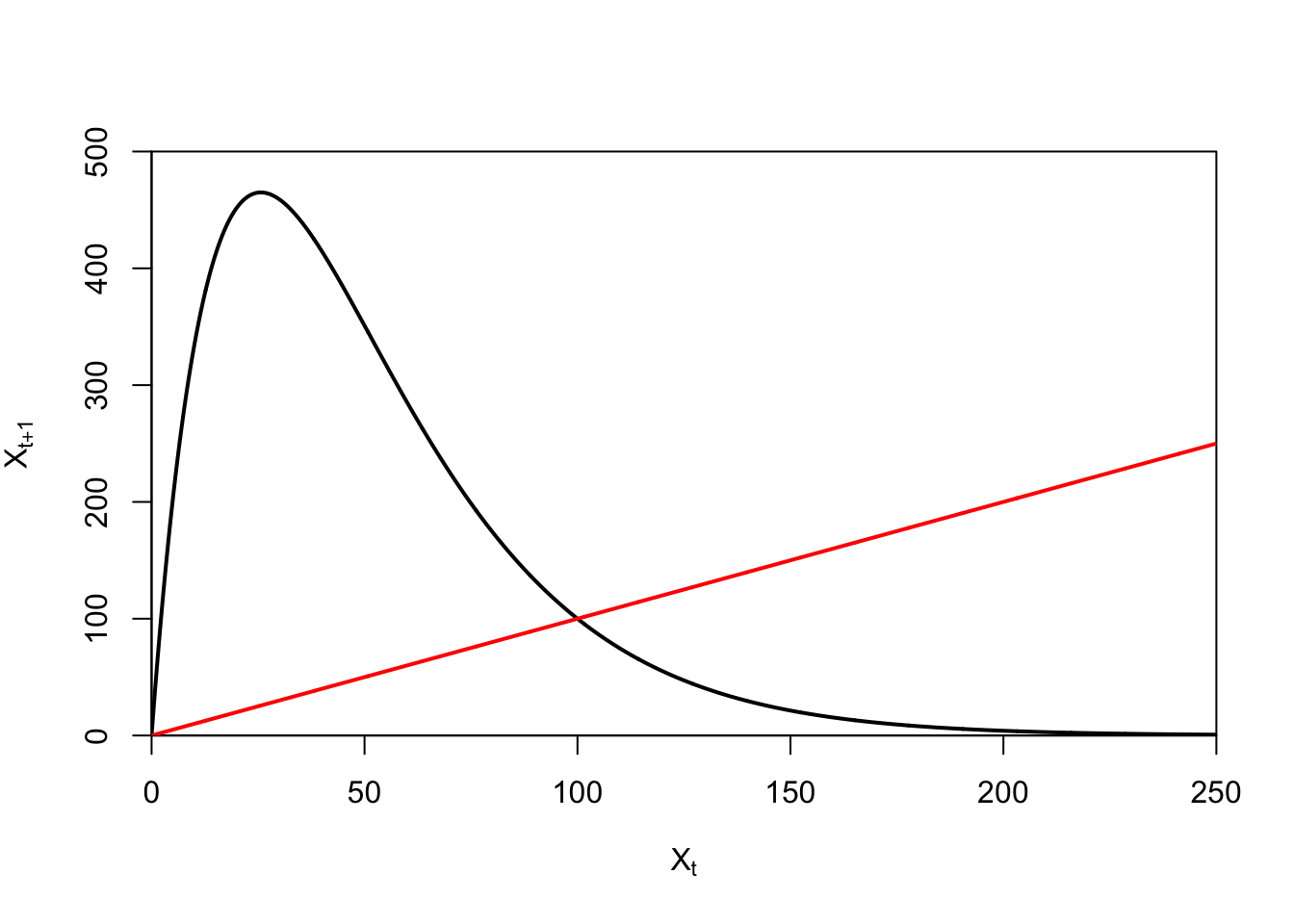
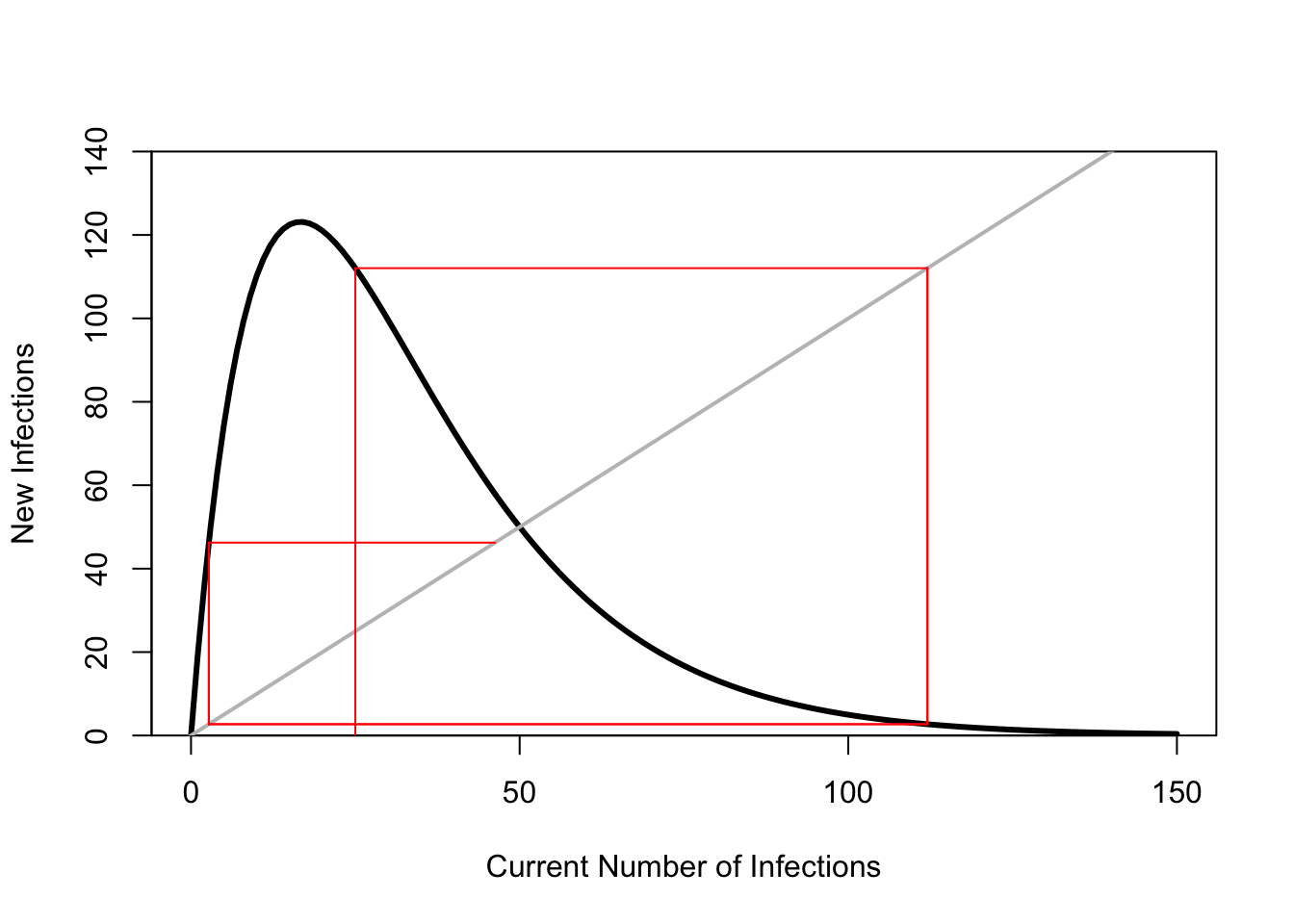
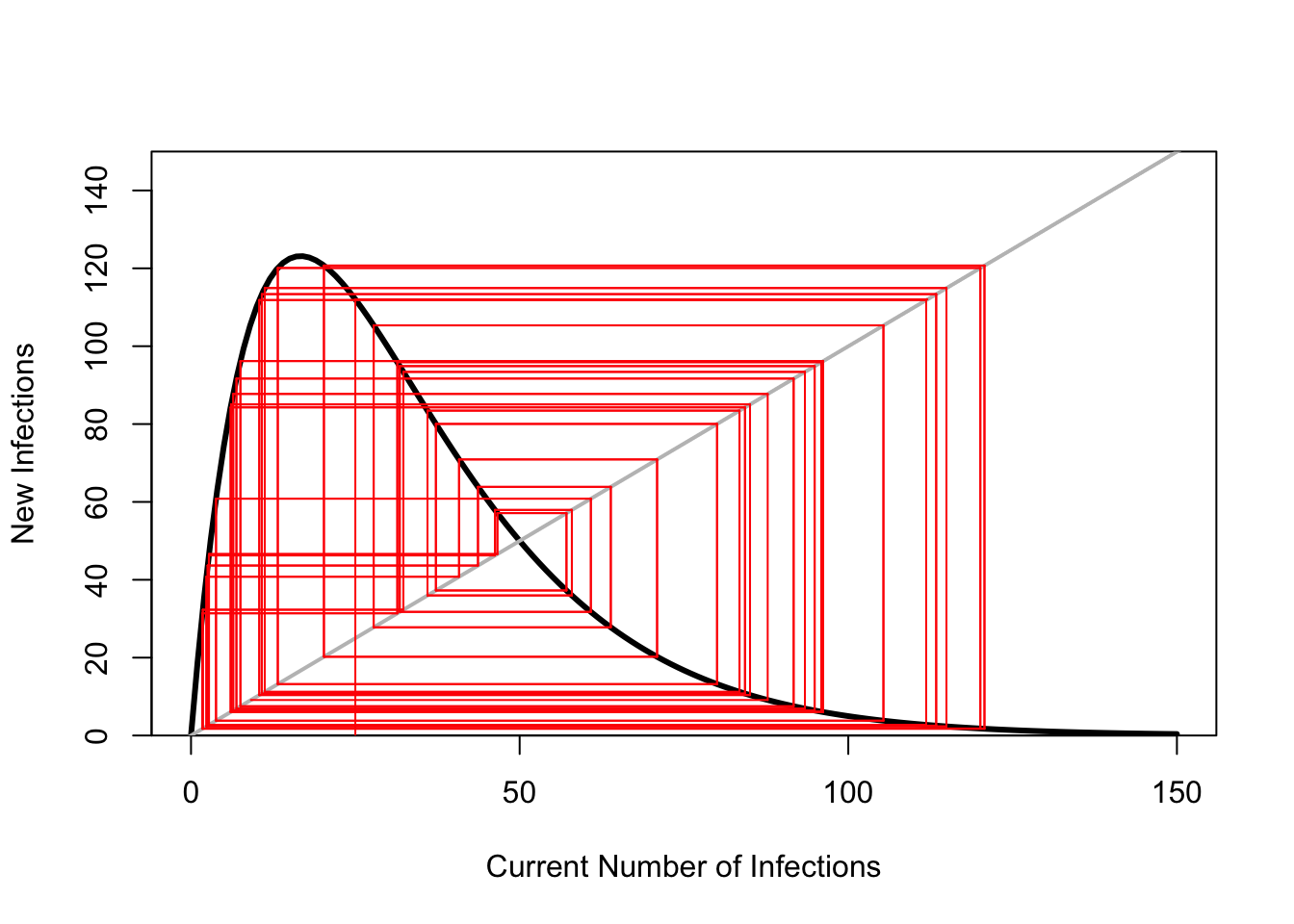
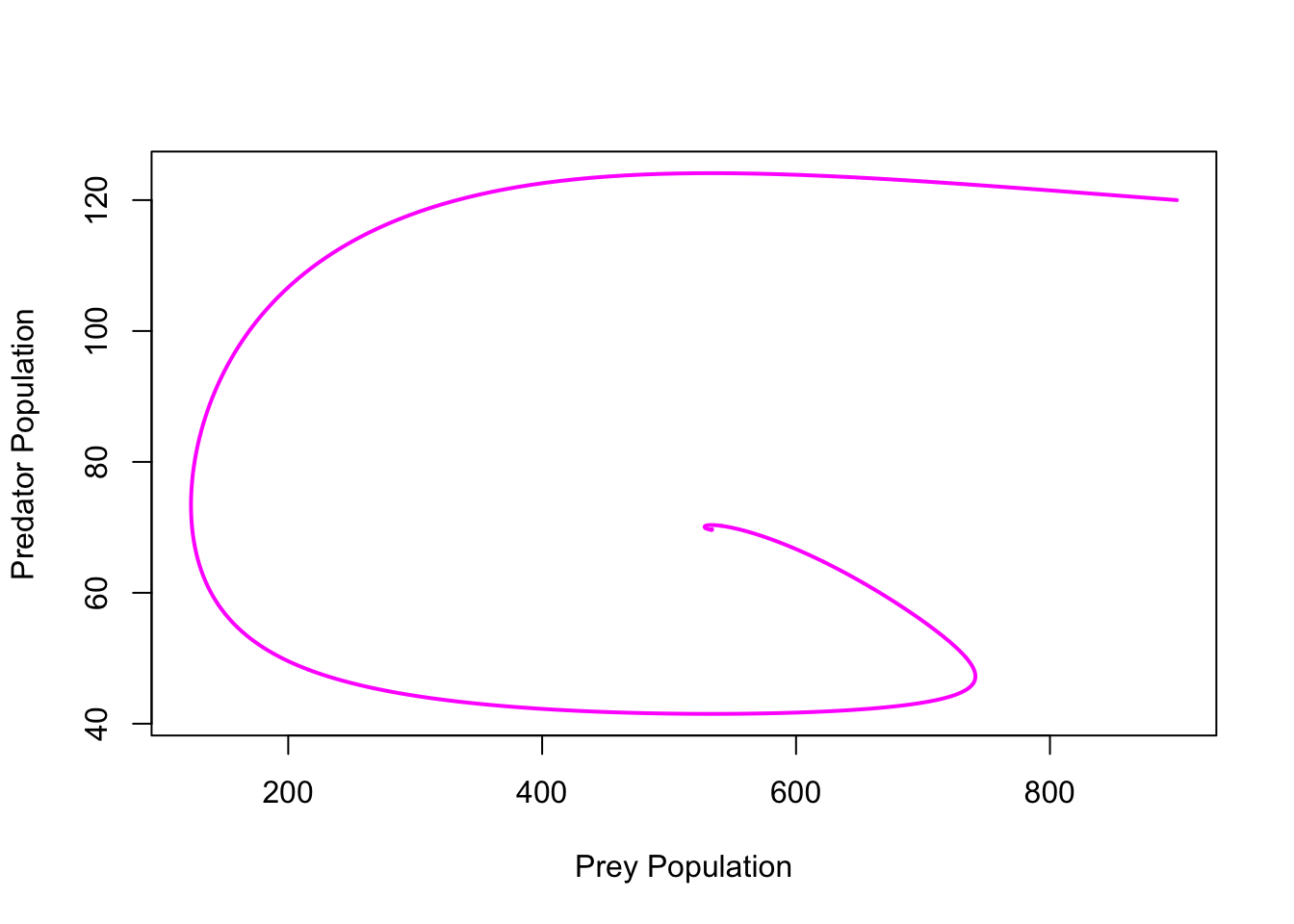
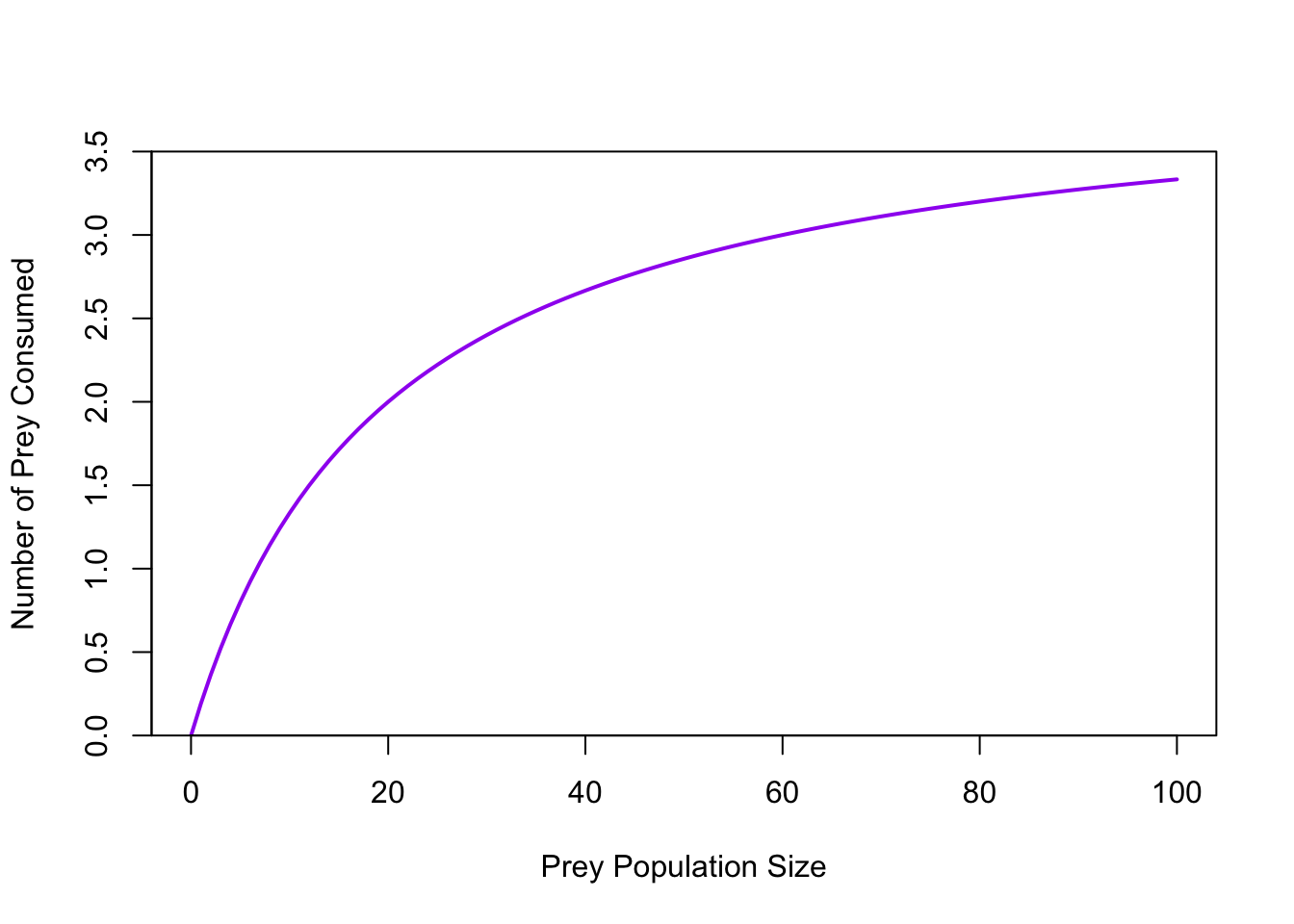
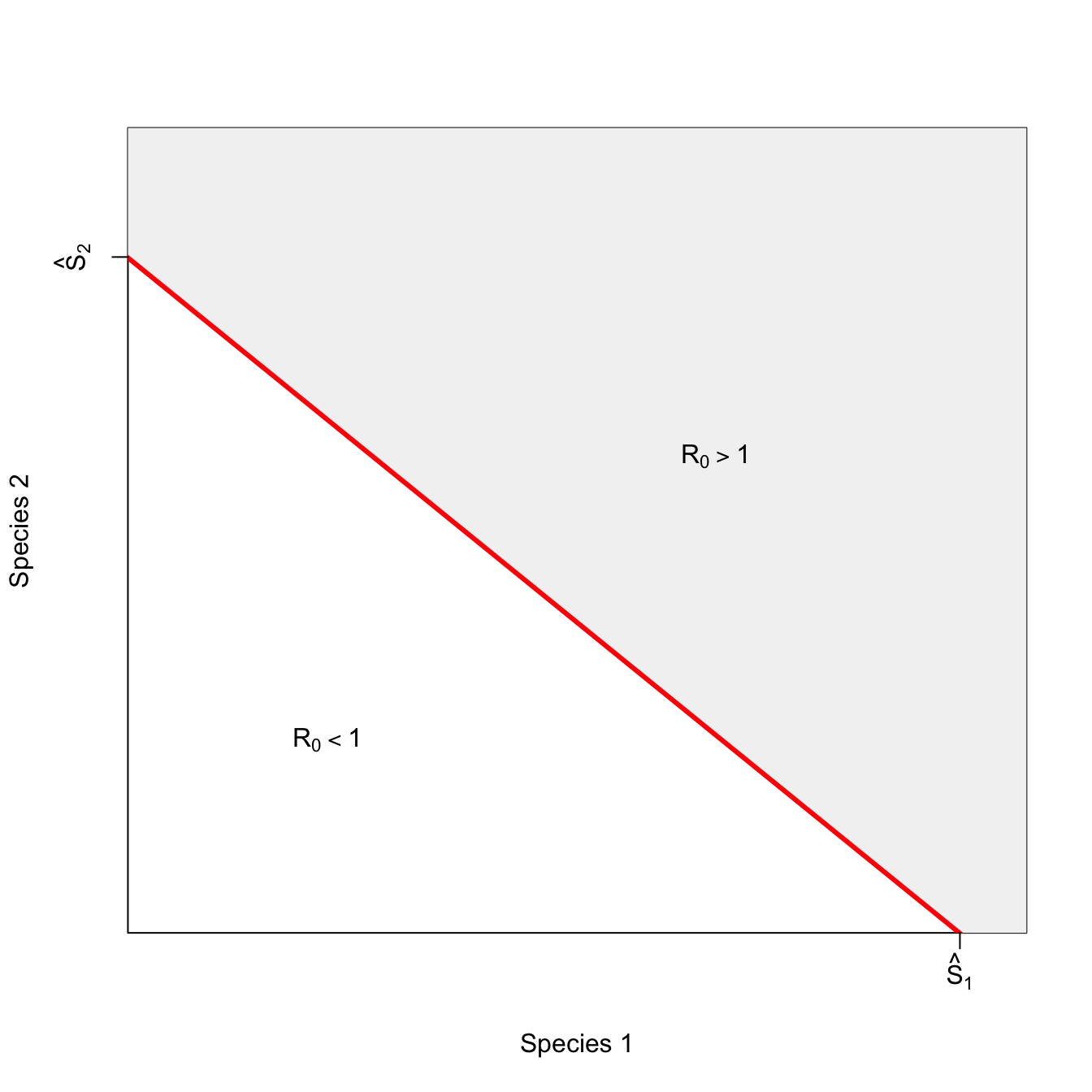
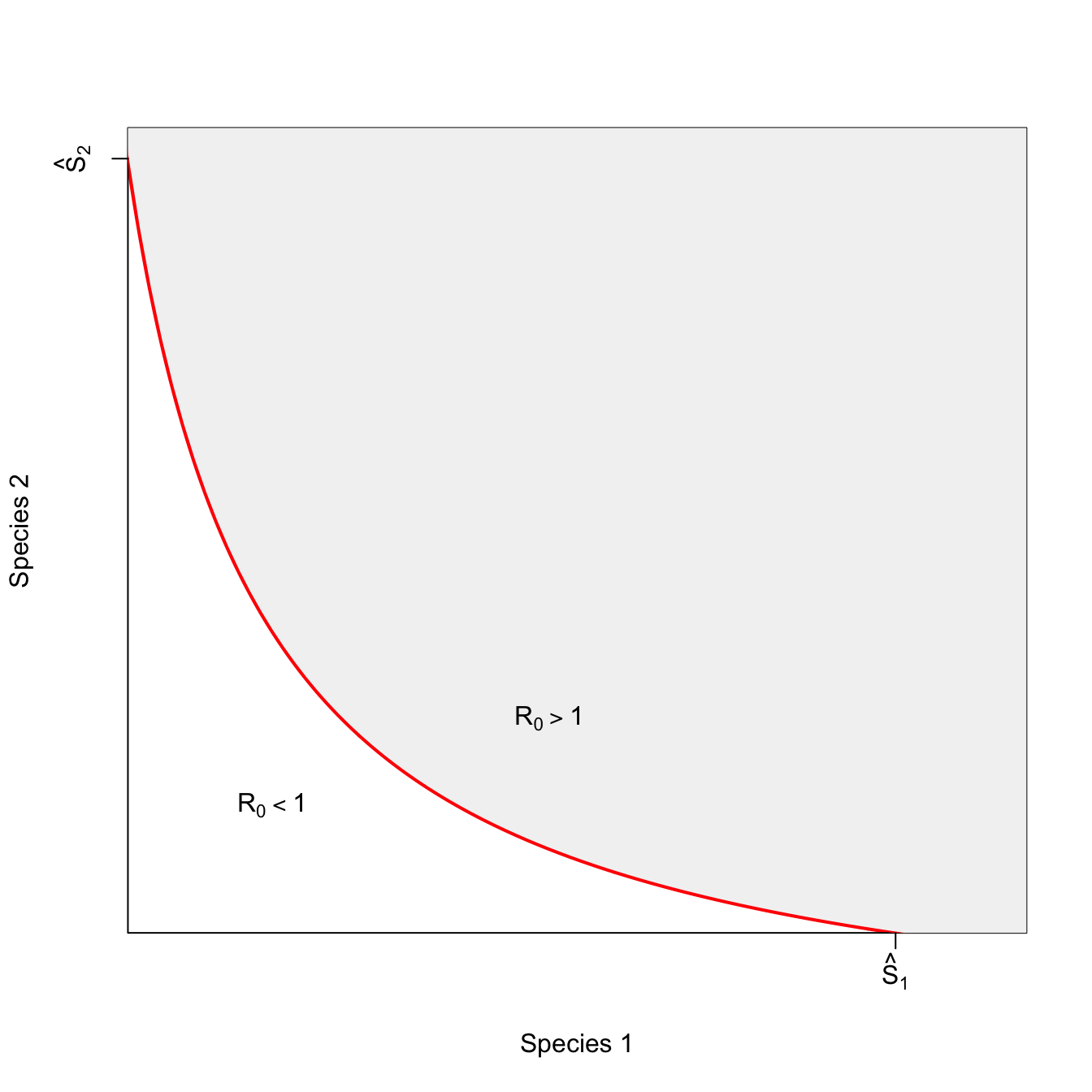
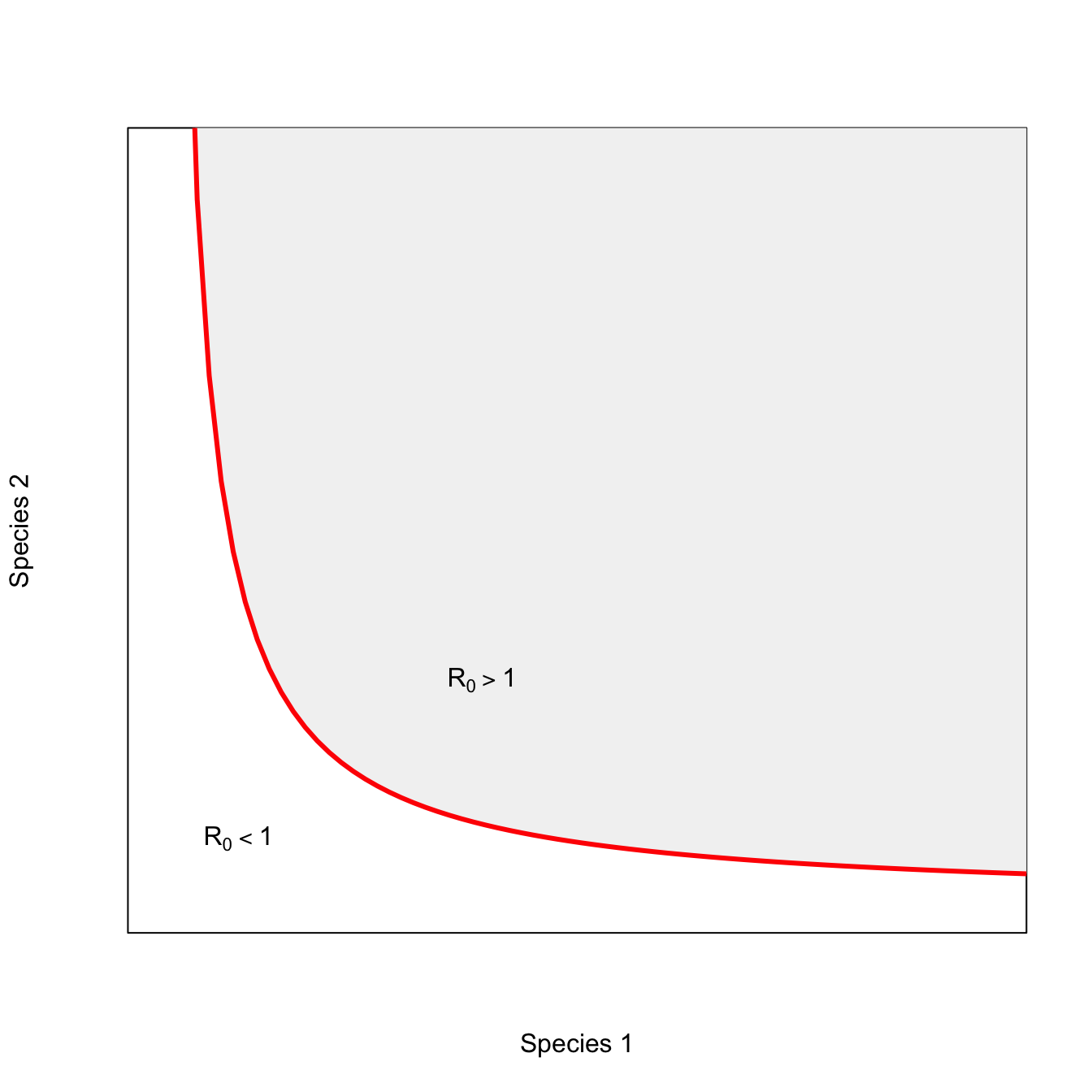
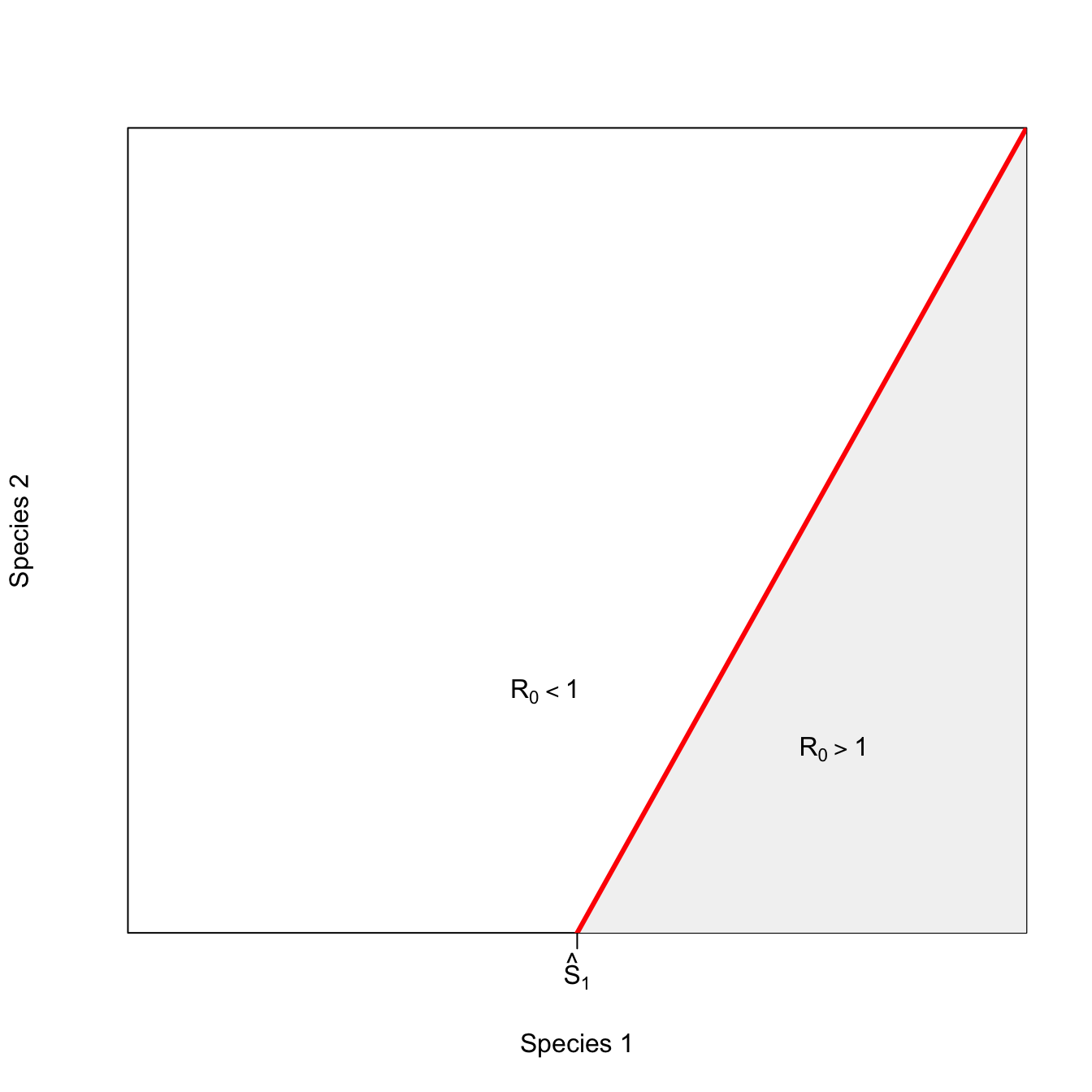
ricker.recruit <- function(r0,K,N) N*exp(r0*(1-(N/K)))
r0 <- 3
K <- 50
N <- 0:150
plot(N,ricker.recruit(r0=r0,K=K,N=N), type="l", col="black", lwd=3, yaxs="i",
ylim=c(0,140),
xlab="Current Number of Infections", ylab="New Infections")
abline(a=0,b=1, lwd=2, col=grey(0.75))
## cobweb -- give it a couple spins around
n0 <- 25
y <- ricker.recruit(r0=r0,K=K,N=n0)
segments(n0,0,n0,y, col="red") #vertical 1
segments(n0,y,y,y, col="red") # horizontal 1
y1 <- ricker.recruit(r0=r0,K=K,N=y)
segments(y,y,y,y1, col="red") #vertical 2
segments(y,y1,y1,y1, col="red") #horizontal 2
y2 <- ricker.recruit(r0=r0,K=K,N=y1)
segments(y1,y1,y1,y2, col="red") #vertical 3
segments(y1,y2,y2,y2, col="red") #horizontal 3
y3 <- ricker.recruit(r0=r0,K=K,N=y)
segments(y,y,y,y3, col="red") #vertical 4
segments(y,y3,y3,y3, col="red") #horizontal 4
y4 <- ricker.recruit(r0=r0,K=K,N=y1)
segments(y1,y1,y1,y4, col="red")